


P02 - Lars Dölken / Florian Erhard
Regulation and Herpes simplex virus 1 counter-regulation of transcriptional bursting kinetics in the early type I interferon response
- Systems biology
- Interferon response
- Herpes simplex virus
- Single cell analysis
- Transcriptional bursts
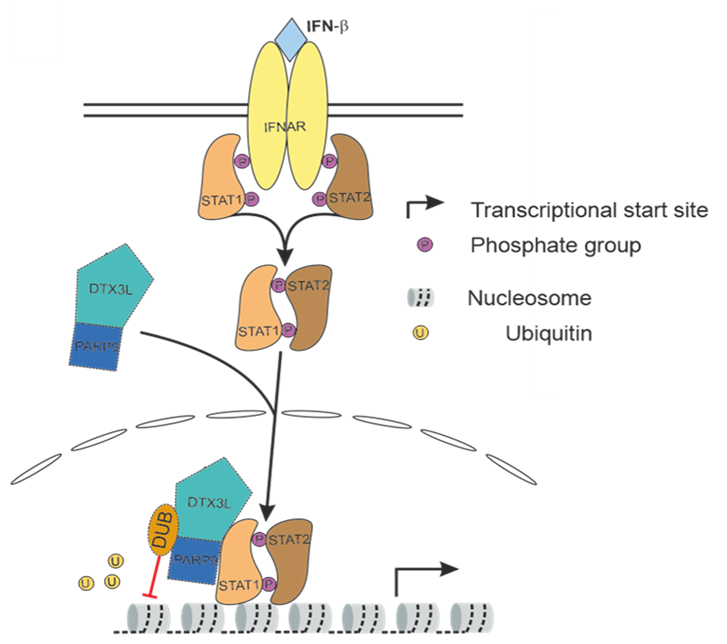
Type I interferons (IFN) induce the expression of hundreds of genes that augment control of invading viruses. Efficient evasion thereof is crucial for productive virus infection and long-term persistence. We discovered that the induction of interferon-stimulated genes (ISGs) in the first two hours of cytomegalovirus infection predominantly results from an increased likelihood of transcriptional bursts. The magnitude of bursts, however, was unchanged compared to basal expression of ISGs in uninfected cells. Recently, the DTX3L/PARP9 ubiquitin ligase complex was shown to bind to the STAT1/STAT2/IRF9 (ISGF3) complex and ubiquitinate histones within ISG promoters, thereby enhancing the expression of a large subset of ISGs. Interestingly, we found that the large HSV 1 tegument protein pUL36 targets DTX3L/PARP9 with its N terminal deubiquitinating (DUB) domain and inhibits two DTX3L/PARP9-attributed ubiquitin-mediated functions, namely the induction of ISGs, and the recruitment of p53 binding protein 1 (53BP1) to sites of DNA damage. We hypothesize that (i) DTX3L/PARP9, which is recruited to ISG promoters via the ISGF3 complex, facilitates switching ISG promoters from a non-permissive to a permissive state and thereby induces more frequent transcriptional bursts, and (ii) that the HSV-1 pUL36 DUB counteracts this to augment productive infection. The main objective of this project is to elucidate the mechanistic role of the pUL36 DUB on the DXT3L/PARP9-ISGF3 interaction in governing the gene-specific transcriptional output of ISGs. The obtained data will illuminate an exciting new cellular mechanism by which ISGs are regulated at the level of transcriptional bursting and how HSV-1 interferes with it. Finally, we will employ our scSLAM-Seq approach and develop the required computational framework to study ISG bursting kinetics at single cell level. In summary, we will study viral manipulation of ISG induction to elucidate novel cellular mechanisms that govern ISG bursting kinetics at chromatin level.
To answer these questions we are applying multiple tools including chromatin immunoprecipitation sequencing (ChIP-Seq), RNA-Seq, genomic manipulation, and sophisticated imaging analysis in appropriate infection models.
This broad spectrum of techniques is only possible thanks to the close cooperation within DEEP-DV.