
P01 - Melanie Brinkmann
Mechanistic insights into the interferon-stimulated gene product zinc finger antiviral protein during lytic and latent human cytomegalovirus infection

- Innate Immune Response
- Human Cytomegalovirus (HCMV)
- The Interferon-Stimulated Gene Zinc Finger Antiviral Protein (ZAP)
- The role of ZAP during lytic and latent HCMV infection
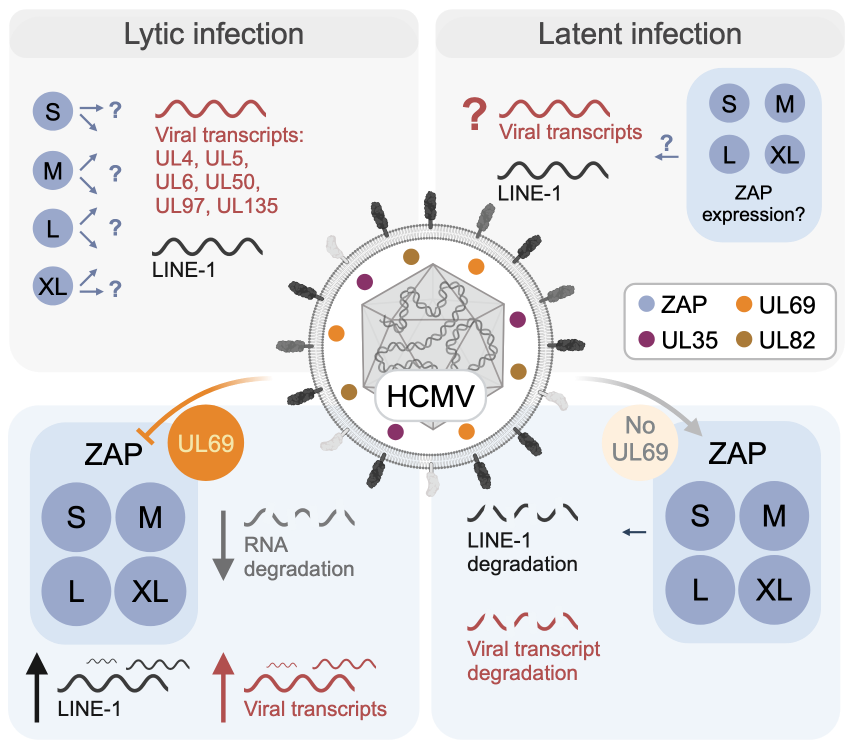
Pattern recognition receptors sense viral infections upon viral entry into the host cell. Subsequently, they induce type I interferon (IFN) and interferon-stimulated gene (ISG) expression to protect the infected cell. The expression profiles and the functions of the more than 300 ISGs known to date vary depending on the virus and cell type. Here, we focus on the relationship between the ISG zinc finger antiviral protein (ZAP), an RNA-binding protein, and the herpesvirus Human Cytomegalovirus (HCMV).
HCMV replicates lytically in certain cell types, such as fibroblasts. In a lytic infection, all viral genes are expressed, the viral genome replicates, new virus particles are assembled, and the new viruses then leave the cell to infect further susceptible cells. A hallmark of herpesviruses is the establishment of a latent infection, which is characterised by very limited viral gene expression and absence of de novo virus particle formation. This form of infection occurs in cells of the myeloid lineage in the case of HCMV.
In this project, we are investigating and comparing the role of ZAP in lytic and latent HCMV infection. We are investigating the importance of the four isoforms of the ZAP protein, ZAP-S/-M/-L/-XL, during the early phase of lytic HCMV infection and infection with other DNA viruses that are the focus of the DEEP-DV consortium: the herpesviruses HSV-1 and KSHV, adenovirus, and BK polyomavirus. Another goal is to find out whether HCMV inhibits the function of ZAP or uses it to its advantage. We address the role of ZAP during latent HCMV infection with a latency model based on induced pluripotent stem cells. For these comparative studies, we use harmonized protocols and reagents as well as state-of-the-art bioinformatic analyses of the DEEP-DV Atlas (Z01). With this integrative approach we aim to identify common and specific mechanisms that are pivotal for the susceptibility of the host and set the path for a productive infection or long-term persistence/latency.